
Pour qu’une molécule puisse avoir de l’activité, il faut d’abord qu’elle puisse avoir de l’affinité pour la cavité liée à l’activité biologique étudiée. Mais cela ne suffit pas, il faut que la molécule puisse trouver un chemin depuis la surface vers cette cavité. Pour les cavités ouvertes en surface, le problème est plus simple mais dès que la cavité est en profondeur cela se complique.
La présente méthode consiste à utiliser un outil de docking comme SMINA et de le détourner pour générer suffisamment de conformations autour de la cavité (distance de 12A par exemple)afin de rechercher un chemin permettant de partir le plus loin possible pour atteindre un des conformations les plus stables.
L’algorithme
Ainsi l’algorithme proposé est le suivant:
– Générer de conformations tant que le nombre de nouvelles conformations est supérieur à une valeur fixée (10% par exemple). L’utilisation d’un LTbox (liste de box définissant le site docking) centré sur le ligand et de pas 3A peut être pertinent.
– Les conformations sont triées par ordre croissant d’affinité.
– Générer un graphe à partir de ces conformations en utilisant une distance maximale de 4,0A( 4.0 est la valeur maximale acceptée pour passer d’une conformation stable à une autre )
– Générer un graphe dirigé (Gxd) par partir du précédent en supprimant toutes les arêtes où la variation d’efficiency est supérieure à 1/nat (nombre d’atomes lourds) et en affectant un poids à chacune des arêtes en utilisant une somme des carrés de l’efficiency et du rmsd (on veut favoriser les chemin de faible saut d’efficiency et de rmsd).
– Pour une conformation finale donnée ( celle d’index 0 par exemple), rechercher toutes les conformations pouvant être atteintes par ce noeuds dans Gxd et les trier par ordre décroissant de distance entre leur centre de masse et celui de la conformation finale.
– Vérifier que ces conformations sont proches des atomes de surface et générer un chemin qui mimimise le poids totale le long du chemin.
# ------------------------------------------------
# Définition des atomes en surface et à l'intérieurde la protéine.
os.chdir(pdb2_site)
Mp_prot_out, Mp_prot_in = get_Mp_surface_from_pdb_prot(return_both=True)
# --------------------------------------------------
# Définition d'un liste de sites de docking.
os.chdir(pdb2_site)
pc_lig = get_pc_from_mol("ligand.sdf")
n=2
N=2*n+1
k = 4
Vx = np.array([k,0,0])
Vy = np.array([0,k,0])
Vz = np.array([0,0,k])
LTbox = [(pc_lig+(-n+i)*Vx + (-n+j)*Vy+(-n+k)*Vz,
15,15,15) for i in range(N) for j in range(N) for k in range(N)]
#---------------------------------------------------
set_log("WARNING")
nat = get_nat_from_mol(mol)
clear_memory()
res = dock_mol_with_smina_until_min_rate(
mol,
cmd_smina=cmd_smina,
nconf=None,
fname="T",
nres_max=300,
rmsd_min_for_clustering=1,
dV_max=0.3*nat,
#dmax_from_Mp_site=12,
nmax_minimize_iters=2,
rmin_new_res=0.2,
exhaustiveness=8,
#Lmol_docked_all=Lmol_docked,
nmax_try=1,
vmax_efficiency=0,
LTbox=LTbox,
nLmol_docked_max=10_000,
#------------------------
pm_processes=6,
pm_parallel=True,
pm_pbar=True,
)
# ------------------------------------------
if res is not None:
Lmol_docked = res
# ------------------------------------------
Lmol_docked = get_Lmol_sorted_by_prop(
Lmol_docked,
prop="minimizedAffinity",
)
# ------------------------------------------
print("get_Lc_rmsd_from_Lmol_same")
Lcv = get_Lc_rmsd_from_Lmol_same(
Lmol_docked,
rmsd_max=4.75,
return_Lcv=True,
K=10,
)
Gx = get_Gx_from_Lcv(
Lcv,
Lattribute="rmsd",
)
# ------------------------------------------
Dc_rmsd = nx.get_edge_attributes(Gx, "rmsd")
#Dc_rmsd = get_D_sorted_by_value(Dc_rmsd, reverse=False)
Dc_rmsd = {tuple(sorted(list(c))):v for c, v in Dc_rmsd.items()}
# ------------------------------------------
print("Création de Gxd")
xdv=1/nat
Gxd = Gx.to_directed()
for c in list(Gx.edges()):
v0 = float(Lmol_docked[c[0]].GetProp("efficiency"))
v1 = float(Lmol_docked[c[1]].GetProp("efficiency"))
dv=abs(v1-v0)
# ------------------------
c2=tuple(sorted(list(c)))
rmsd = Dc_rmsd[c2]
# ------------------------
dv2 = pow(dv/xdv,2)
rmsd2 = pow(rmsd/15,2)
w = dv2 + rmsd2
Gxd[c[1]][c[0]]["weight"]= w
Gxd[c[0]][c[1]]["weight"]= w
if v1 > v0 + xdv :
Gxd.remove_edge(c[0], c[1])
elif v1 > v0:
Gxd[c[0]][c[1]]["weight"]= w*2
if v0 > v1 + xdv :
Gxd.remove_edge(c[1], c[0])
elif v0 > v1:
Gxd[c[1]][c[0]]["weight"]= w*2
print("Done")
#-----------------------------
# A partir des conformations les plus stables,
# recherche des conformations connectées à celle-ci dans Gxd
# La conformation finale (n0) doit être proche (d<9A) du ligand.
n0=-1
while True:
print(n0, end="\r")
n0+=1
pc0 = get_pc_from_mol(Lmol_docked[n0])
Sn_all = {n0}
Sn_new = set(Gxd.predecessors(n0))
#LSn_new = [Sn_new]
while True:
Sn2 = set()
for n in Sn_new:
Sn2 |= set(Gxd.predecessors(n))
Sn_all |= Sn_new
Sn_new_2 = Sn2 - Sn_all
Sn_new = Sn_new_2
if len(Sn_new) == 0:
break
#LSn_new.append(Sn_new)
Ln1 = sorted(list(Sn_all))
Mpc = get_Mpc_from_Lmol(
[Lmol_docked[i] for i in Ln1])
Vd = get_Vdist_p_Mp(pc0, Mpc)
dmax = np.max(Vd)
if dmax < 4.0:
continue
print(n0, dmax)
LT = list(c for c in enumerate(Vd.tolist()))
LT = get_LT_sorted(LT, 1, reverse=True)
Li2 = [T[0] for T in LT]
Ln2 = [Ln1[i2] for i2 in Li2]
v0 = float(Lmol_docked[Ln2[0]].GetProp("efficiency"))
v1 = float(Lmol_docked[Ln2[-1]].GetProp("efficiency"))
if v0 < v1:
print("No solution")
break
In2 = iter(Ln2)
V = np.array([T[1] for T in LT])
show_LV(
[V],
xlabel="index of the conformations",
ylabel="distance bw center of mass",
ymax=25,
ymin=0,
)
print("found")
break
#--------------------------------------
# A partir des conformations les plus éloignées,
# Recherche de chemin qui commencent par une conformation en surface
# de la protéine.
for ns in In2:
Mp_conf = get_Mp_from_mol(Lmol_docked[ns])
Mc_out = get_Mc_bw_Mp(Mp_prot_out, Mp_conf, dmax=4.5)
Mc_in = get_Mc_bw_Mp(Mp_prot_in, Mp_conf, dmax=4.5)
if (Mc_out is None) or (Mc_in is None):
continue
if len(Mc_out) >= len(Mc_in):
print("OK, c'est une conformation en surface")
print(ns)
breakOn utilisera les abbréviations suivantes:
conf_fin pour conformation finale
conf_init pour la première conformation du chemin qui doit être proche ou sur la surface.
Définition des atomes de surface pour la protéine.
Pour obtenir les atomes qui définissent la surface de la protéine, l’algorithme de Delaunay a été appliqué et les sphères de rayon de plus de 6A ont été sélectionnées. Les atomes qui forment la surface de la protéine s’en déduisent.
Cas de la cible CHEMBL4980 , récépteur acétylcholine alpha 7
Les essais suivants ont été réalisés avec le PDB 7KOQ.
Exemples de molécules qui fonctionnent
Ex. 1

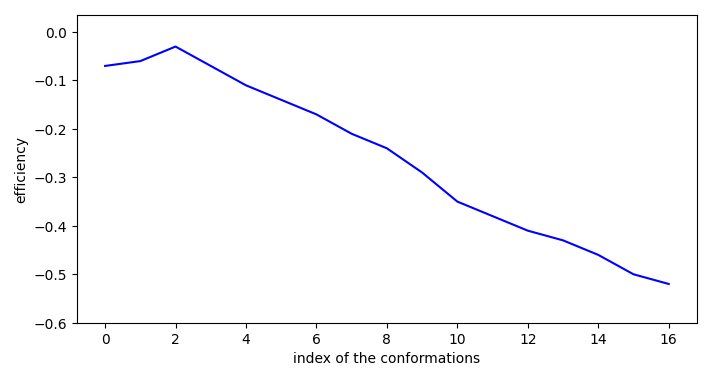
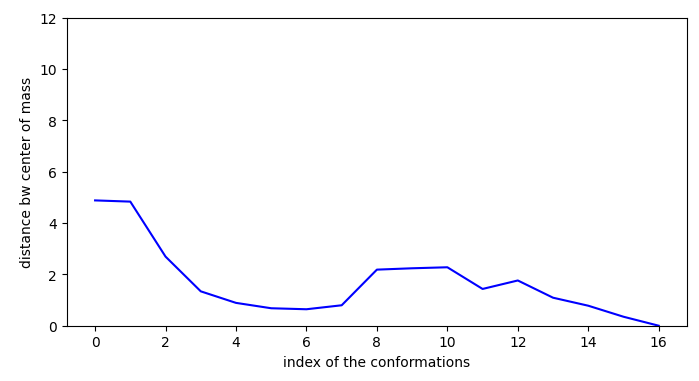
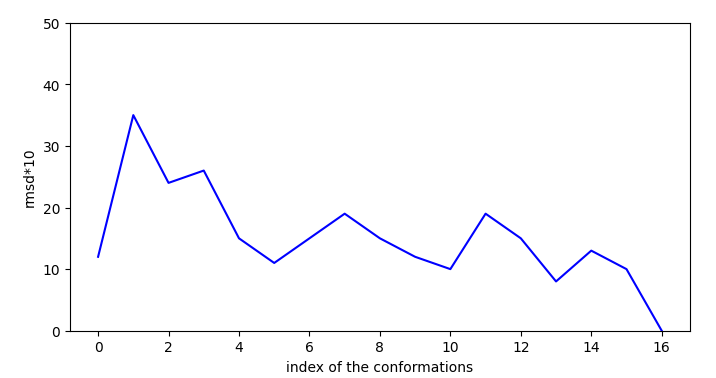
L’idéal est donc d’avoir un accès à une conformation en surface proche qui permettent une certaine stabilisation (ici -0.05 en efficiency) et pas (ou presque) d’activation.
Ex.2
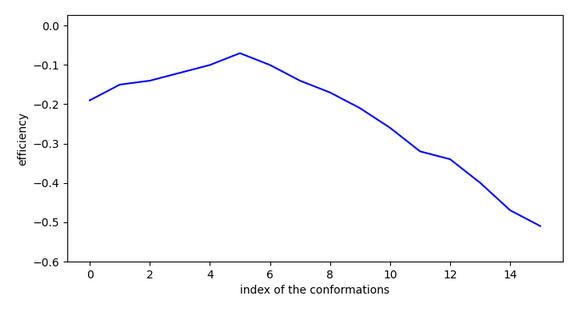
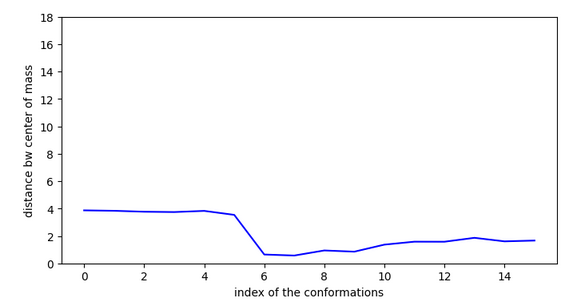
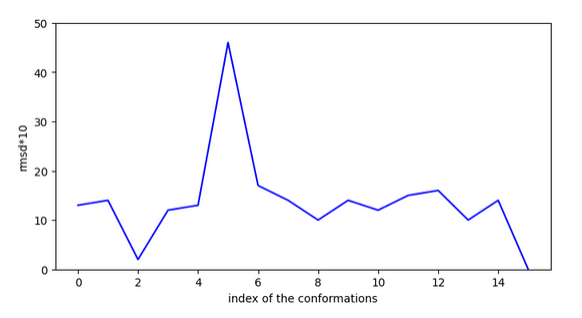
Cette molécule fonctionne mais moins bien que la précédente car il y a une activation et un saut de 4,5A entre 2 conformations au moment de l’activation. Un rmsd max. entre conformations de 4,75A est donc nécessaire pour vaider cette molécule. Un modélisation dynamique doit alors finaliser cette validation.
Ex. 3
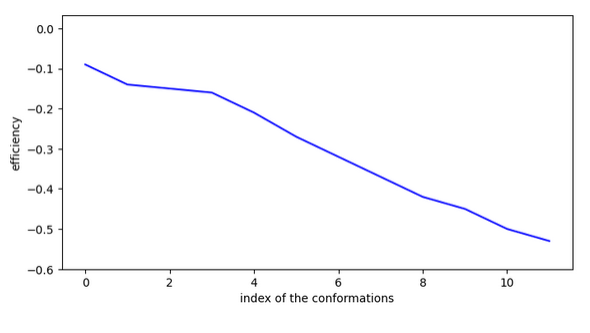
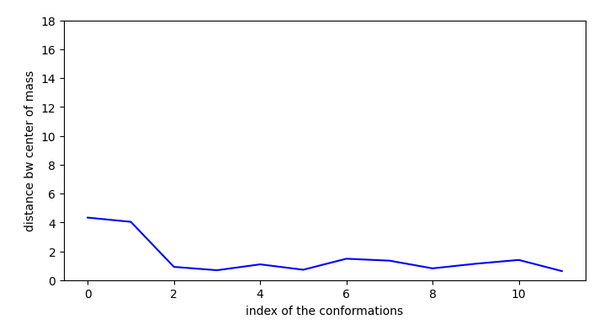
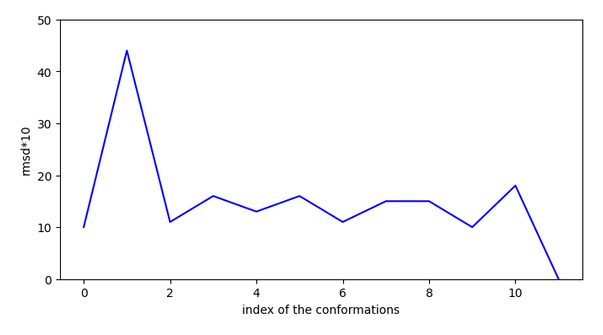
Cette molécule est comme la précédente, elle doit réaliser un saut de plus de 4A mais à priori il n’y a pas d’activation nécessaire sur ce chemin.
Ex. 4
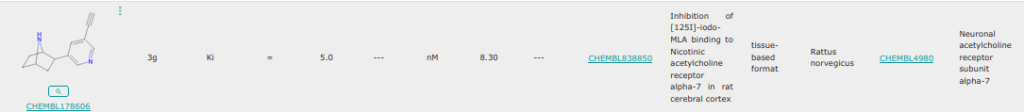
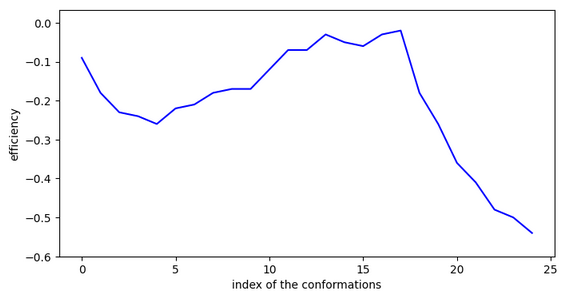
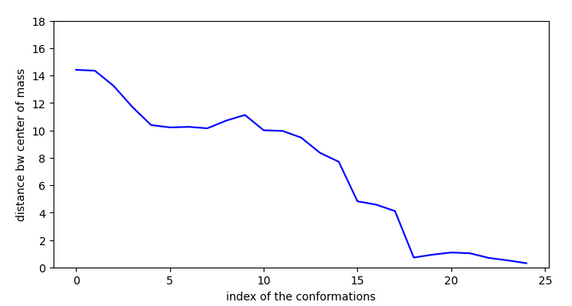
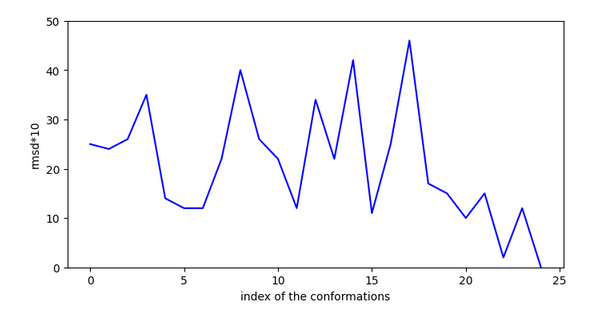
Cette molécule possède un pKi plus élevé que les autres car elles peut se stabiliser en surface et accéder à l’entrée de la cavité par des sauts de moins de 4,5A en rmsd.
Ex. 5

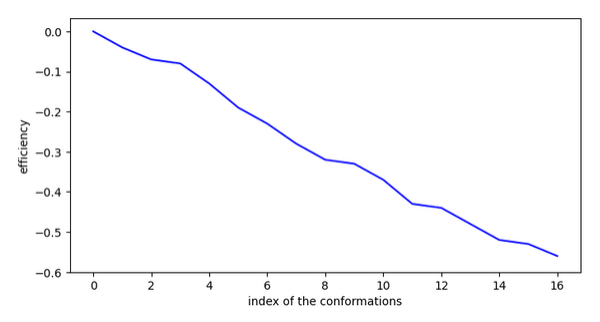
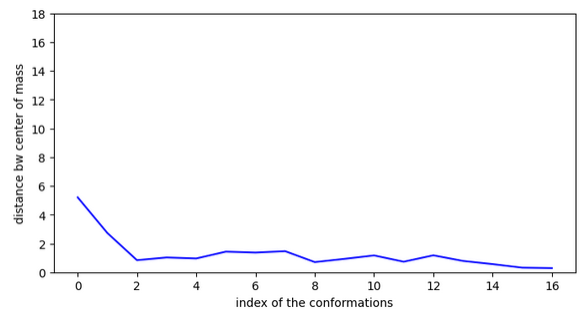
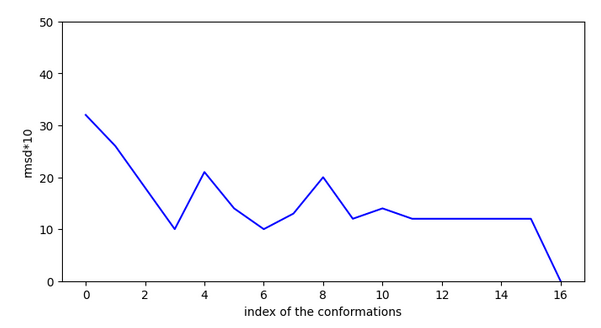
Ex.6 pKi=10,6
O=S1(=O)c2ccccc2-c2ccc(N3CCN4CCC3CC4)cc21
A priori, cette molécule ne trouve pas de chemin pour rentrer dans le site. Elle devrait plutôt agir en bloquant l’entrée de la cavité.
Exemples de molécules qui fonctionnent moyennement
Ces molécules ont un pKi entre 5,5 et 6
Ex. 1
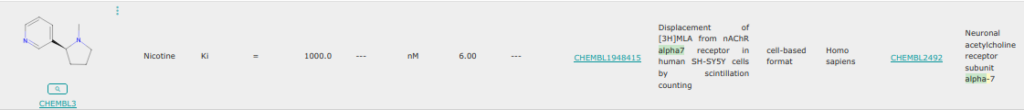
La nicotine possède un pKi de 6,0 pour le récépteur acétylcholine alpha 7 chez l’homme ce qui est une valeur moyenne. A priori cette molécule n’a pas assez d’affinité en surface pour bien rentrer dans le site (efficiency > -0.1).
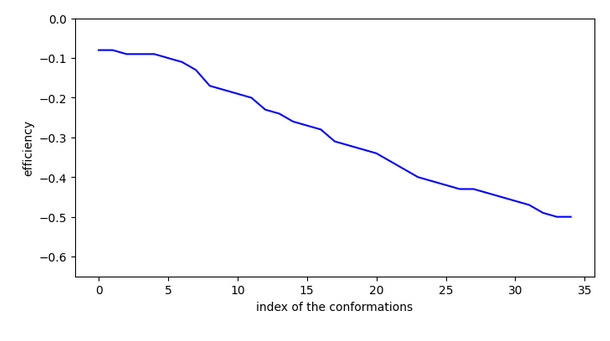
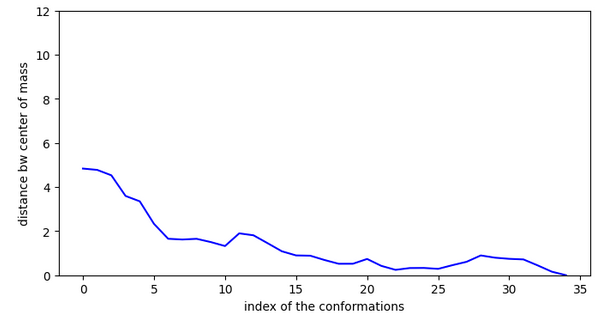
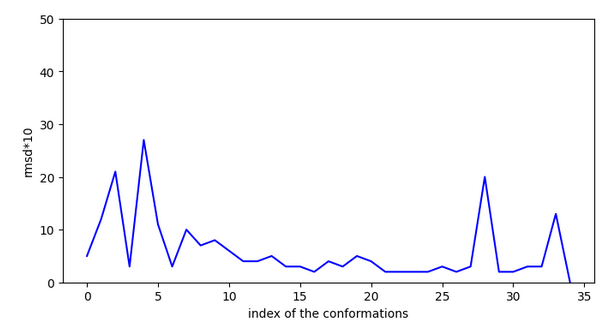
Ex. 2

Cette molécule a une forte affinité mais elle doit commencer son chemin à une distance de 12A. Sa stabilisation en surface est très bonne mais il y a une activation de 0,1 en efficiency. A priori, cette distance est trop important et l’activivation également.
Exemples de molécules qui ne fonctionnent pas
Ces molécules ont un pKi inférieur à 5
Ex. 1

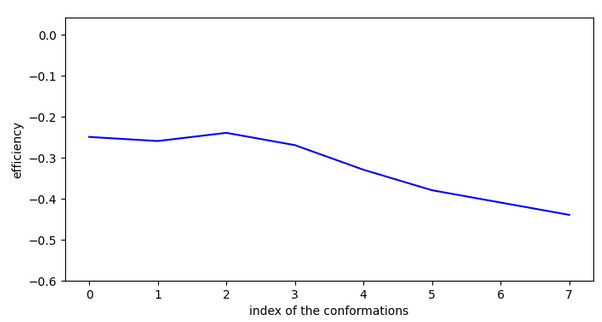
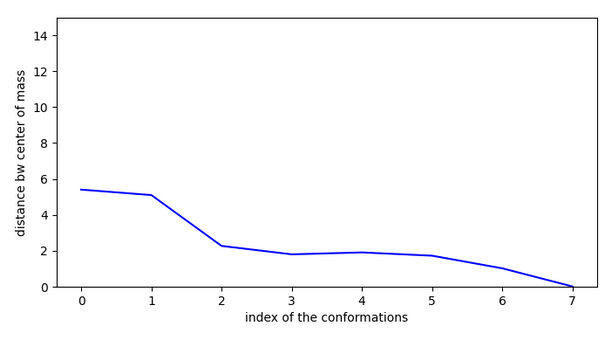
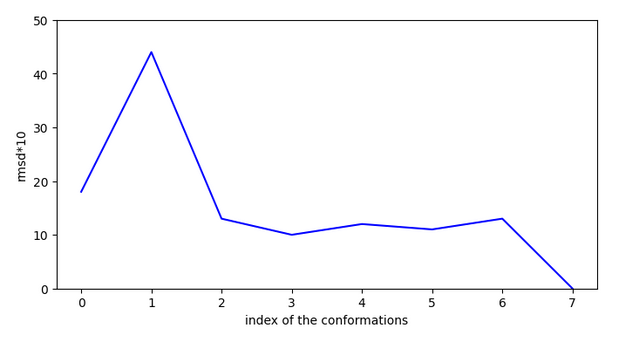
La molécule CHEMBL61616 n’est pas active sur la cible CHEMBL4980 , récépteur acétylcholine alpha 7.
L’explication que l’on peut donner est qu’elle se stabilise fortement et doit faire un saut de plus de 4A. A priori, les grands sauts sont permis uniquement lorsque le ligand possède une efficiency < -0.1.
Ex. 2
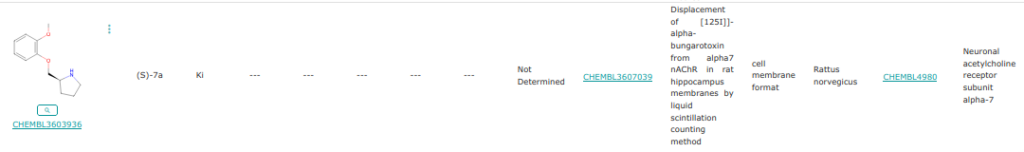
Les conformations de départ qui mênent à la cavité ne sont pas prêts de la surface et le chemin est court.
Les conformations de surface conduisent à une autre conformation finale.
Exemples intéressants
Ex. 1
Pour un méthyl en plus, ça marche mieux !
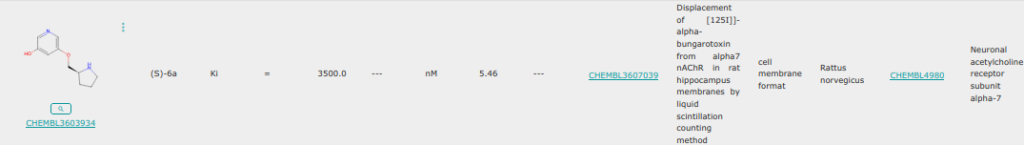
L’énatiomère Oc1cccc(OC[C@@H]2CCCN2)c1 ne trouve pas facilement de chemin.
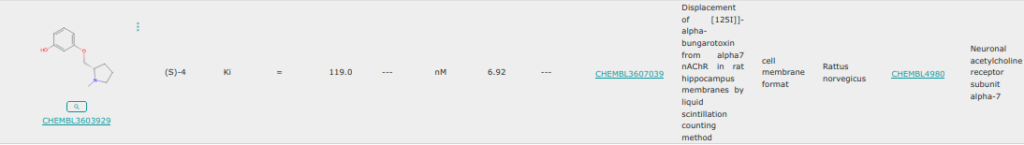
Je n’ai pas pu obtenir de résultat pour cette molécule ! J’ai essayé avec l’autre énantiomère et je n’ai pas pu trouvé de chemin également. Donc ce résultat est cohérent avec le résultat obtenu sans le méthyl, en effet, ajouter cet élément à une molécule avec un pKi faible peut difficilement permettre d’obtenir un pKi plus grand même si des contre-exemples doivent exister pour des cavités en surface par exemple.